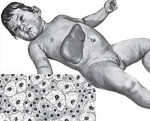
Описание болезни Липоидозы

Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
Липоидозы
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
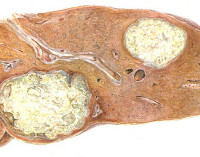
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы – жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название – болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы – плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов – сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
| Литература1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. и др.// Медицинский совет. — 2016 — №1.2. Клиническая генетика/ Давиденкова Е.Ф., Либерман И.С. — 1975.3. Наследственные нарушения обмена липидов: учебно-методическое пособие/ Якимович Н.И. — 2016. | Код МКБ-10E75 |